ARTÍCULO DE REVISIÓN
Nutrición y genoma
Nutrition and Genome
Tamara Rubio González
Universidad Médica de Santiago de Cuba, Cuba.
RESUMEN
El estado de salud del hombre es el resultado de la interacción entre su individualidad y el entorno. Las diferentes respuestas de los individuos a una misma dieta han despertado la atención de los investigadores, quienes, además, han observado repercusión fenotípica de algunos esquemas alimentarios en diferentes latitudes. En el siglo XXI, la preocupación por la interacción entre el genoma y la dieta ha permitido el desarrollo de la Genómica nutricional o Nutrición molecular, disciplina que aporta el conocimiento que permite hacer un diagnóstico y establecer un tratamiento nutricional basado en el genotipo individual. Este trabajo constituye una revisión sobre las interrelaciones entre los alimentos y los genes, con el objetivo de propiciar la reflexión sobre un integrante importante de nuestro entorno: la dieta.
Palabras clave: nutrición; Genómica nutricional; dieta.
ABSTRACT
The man's state of health results from the interaction between his individuality and the milieu. Individuals responding differently to the same diet has caught the attention of investigators, who also observed the phenotypic repercussion of some food schemes in different parts of the world. In the twenty-first century, concern for the interaction between genome and diet has permitted the development of nutritional genomics or molecular nutrition, a discipline that provides knowledge permitting diagnosis and to establish a nutrition treatment based of the individual genotype. This work reviews the interrelation between food and genes, with the aim to favor reflection about a critical member of our milieu: diet.
Keywords: nutrition; genomics; diet.
INTRODUCCIÓN
El estado de salud del hombre es el resultado de la interacción entre su individualidad y el entorno. Desde hace algunos años, se ha comenzado a ver la dieta como un factor importante de ese entorno y se ha pensado en ella como desencadenante de enfermedades. Las variaciones genéticas individuales pueden determinar diferencias en la asimilación, metabolismo, almacenamiento o excreción de los nutrientes.1,2
Por otro lado, los resultados del Proyecto Genoma Humano y la tecnología desarrollada han permitido encontrar nuevas evidencias del papel de la dieta sobre el genoma y de la repercusión de sus variaciones en las diferentes respuestas observadas. De este modo, ha surgido una nueva disciplina que se dedica a estudiar el efecto de los nutrientes sobre el ADN, la cual recibe el nombre de Genómica nutricional. Paralelamente, el desarrollo de otras "ómicas" como la Proteómica, la Transcriptómica y la Metabolómica ha complementado el conocimiento de esta nueva rama.3,4 En esta revisión pretendemos aproximarnos a los conocimientos que relacionan la nutrición y el genoma, como inicio de un camino en la medicina actual que llevará a la personalización de la dieta.
INTERACCIÓN GEN-AMBIENTE
La individualidad del hombre está dada por variaciones o polimorfismos (SNPs) en el genoma que representan solo el 0,1 % del código genético, pues se ha comprobado que la especie humana tiene similar el 99,9 % del genoma. Las variaciones del genoma son muy diversas; pueden ocurrir en intrones o en exones, y en estos últimos pueden implicar o no cambio de aminoácidos en la cadena polipeptídica resultante. Otra forma de variación son los cambios en el número de copias de un gen, que pueden provocar su sobreexpresión. Independientemente de la modificación de que se trate, esté relacionada directa o indirectamente con una enfermedad, primero hay que dilucidar si estamos en presencia de una relación genotipo-fenotipo directa, o si existe la posibilidad de una modulación o interacción ambiental.4,5
Para algunas enfermedades los factores genéticos tienen mayor relevancia y para otras, los factores ambientales son los preponderantes; pero, tanto en unas como en otras, la interacción genoma-ambiente está presente.4 De los factores ambientales a los que nos exponemos continuamente, la dieta es el factor cuantitativamente más importante, porque siempre estamos expuestos a ella; pero además, es posible adaptarla y modificarla, atendiendo a los requerimientos de cada individuo.
La interacción gen-dieta está suficientemente documentada en la literatura médica, ejemplo de ello son los errores innatos del metabolismo, como la fenilcetonuria y la galactosemia entre otros; en estas enfermedades es posible modificar el fenotipo causado por una mutación genética, mediante una dieta restrictiva.6 Sin embargo, en las enfermedades complejas, esta relación no está bien definida, aunque también es evidente. La obesidad, la hipertensión arterial, la diabetes mellitus y el cáncer, por solo citar algunas de ellas, involucran un grupo de genes y sus mecanismos son complejos, pero el papel del ambiente en ellas, está también abundantemente referido por numerosos investigadores.7-9
El papel de la dieta como factor ambiental en las tres primeras enfermedades ha sido reportado extensamente.8-10 En 1981, Doll y Peto señalan a la dieta como un factor tan importante como el consumo de cigarrillo en el riesgo de padecer cáncer. En 2007, la World Cancer Research Fundation, remarca la importancia de la dieta como un modificador del riesgo de cáncer. En 2012 un estudio EPIC demostró los efectos de la ingesta dietaria en el cáncer.
GENÓMICA NUTRICIONAL
En el siglo XXI, la preocupación por la interacción entre el genoma y la dieta ha permitido el desarrollo de la Genómica nutricional o Nutrición molecular, disciplina que aporta el conocimiento que permite hacer un diagnóstico y establecer un tratamiento nutricional basado en el genotipo individual, mediante dos ramas principales: Nutrigenética y Nutrigenómica.11-13 La Nutrigenética estudia las diferentes respuestas fenotípicas de los individuos a los nutrientes, en dependencia del genotipo de cada cual. La Nutrigenómica analiza los efectos de los nutrientes sobre la expresión de los genes y la interrelación de estos cambios con el proteoma y el metaboloma.12,14
NUTRIGENÉTICA
Esta rama estudia las diferentes respuestas fenotípicas a la dieta, según el genotipo.12,14 Justifica por qué los individuos responden de manera diferente a una misma dieta. La respuesta a un nutriente dependerá de las variaciones genéticas de cada individuo; dependiendo de ello cada persona va a absorber, metabolizar o utilizar los nutrientes de diferentes maneras: algunos serán normorrespondedores, otros hiporrespondedores y hay un tercer grupo de hiperrespondedores; pero en cada grupo, habrá una gama de respuestas posibles, dependiendo del genotipo individual.12
En el caso de la obesidad, a partir de sus manifestaciones monogénicas se ha conseguido identificar genes clave, que no solo producen obesidad severa por mutaciones (como en los síndromes Prader-Willi, Alström y Bardet-Bield); también se ha logrado identificar en ellos variaciones que afectan menos su función, y que están presentes con elevada frecuencia en la población contribuyendo, de cierta forma, a incrementar el riesgo de obesidad; entre ellos se destacan el gen de la leptina (LEP) y el gen del receptor de la leptina (LEPR).15,16
El gen LEP se localiza en 7q31.3; su producto proteico, la leptina envía una señal al cerebro sobre el tamaño del tejido adiposo, por lo que actúa como factor saciante. Los individuos con mutaciones severas de este gen manifiestan una hiperfagia con obesidad extrema desde la infancia, aunque su peso al nacer es normal; en niños algo mayores, lleva a una conducta agresiva especialmente cuando se ven privados de alimentos. Este cuadro está asociado con hipotiroidismo hipotalámico e hipogonadismo hipogonadotrópico.10,16
El gen LEPR tiene locus 1p31; la unión de la leptina al receptor provoca su dimerización y el reclutamiento de enzimas de la familia JAK (del inglés Janus Kinase) que fosforilan al receptor en sitios específicos, los cuales sirven para el reclutamiento de las proteínas de la familia STAT (del inglés Signal Transducer ana Activator of Transcription), que son fosforiladas por las JAK. Las proteínas STAT fosforiladas forman dímeros que son transportados al núcleo celular para unirse a promotores de genes específicos, regulando la transcripción génica.10,15
El gen POMC tiene locus 2p23.3 y se traduce en un largo polipéptido que tiene varias regiones: el dominio amino-terminal, que de acuerdo con su procesamiento puede dar origen a dos péptidos: el g3-MSH (del inglés Melanocite Simulating Hormone) o el g1-MSH; seguidamente hay un péptido de unión, luego la ACTH y termina con la b-LPH (del inglés Lipotropin Hormone). Las mutaciones observadas en el gen pueden suprimir la expresión total de este o modificar específicamente alguno de los péptidos codificados en él. La deficiencia completa de expresión del gen POMC se traduce en bajos niveles de ACTH circulante, por lo que se produce una hipocortisolemia. Como resultado aparece hipoglicemia e ictericia persistente y los individuos afectados son susceptibles a las infecciones. Sin embargo, la hipocortisolemia se acompaña de obesidad y no de pérdida de peso, como es lo habitual, y los pacientes sufren una hiperfagia incontrolable. La ausencia de MSH hace que los pacientes tengan la piel pálida y el pelo rojizo.10,16
Otros genes involucrados en la obesidad son PC1, MC4R, SIM1 y FTO.16,17 Estas mutaciones que producen obesidad monogénica son infrecuentes; los polimorfismos son los que más se encuentran y presentan diferentes prevalencias según las poblaciones; pueden estar asociados a obesidad poligénica o multifactorial. En este tipo de obesidad, la herencia de genes específicos que regulan el apetito (leptina, grelina, receptores de melanocortina, NPY), la termogénesis y el metabolismo energético (ADRB2, ADRB3, UCP´s, etc.) y la adipogénesis (PPAR, PXR, adiponectina, etc) interactúan con la dieta para producir la enfermedad. Las variantes genéticas presentan el mismo comportamiento; los individuos portadores de la mutación Gln24Glu del gen ADRB2, o del polimorfismo Pro12Ala del gen PPARG2, si tienen una ingesta elevada de carbohidratos, poseen mayor riesgo relativo de obesidad.16,17
La hipertensión arterial se ha asociado a la cantidad de angiotensina circulante. Un SNP, denominado AA, en la posición del nucleótido 6 del gen de la angiotensina (AGN) se ha relacionado con el nivel de esta hormona circulante. Alrededor del 60 % de los afroamericanos tienen la variante AA, y el resto son heterocigóticos (AG) para esta posición. Los individuos con el genotipo AA que tienen una dieta hiposódica y baja en carbohidratos muestran una reducción de la tensión arterial, pero esta misma dieta resulta menos efectiva en la reducción de las cifras tensionales en individuos con el genotipo GG. En el gen AGN se han descrito 15 variantes alélicas relacionadas con la hipertensión arterial.4,9
Otro gen que se ha podido asociar a la ingesta de sal y la hipertensión es ADD1, con locus 4p; el cual codifica una proteína llamada aducina 1, que se localiza en el citoesqueleto de membrana y que favorece la unión entre la espectrina y la actina. La aducina 1 tiene una importante función en la arquitectura de la membrana, debido a su interacción entre los filamentos de actina y de espectrina y, potencialmente, sobre la actividad de ciertos canales, en particular el cotransporte de Na-K-Cl y la Na-KATPasa. El polimorfismo G460W del gen ADD1 es más frecuente en los hipertensos que en los normotensos, y parece predisponer a una sensibilidad particular a la sal y a la hipertensión.18
Las enfermedades cardiovasculares constituyen un ejemplo clásico de como el genotipo determina la respuesta a los nutrientes. El gen APOE, localizado en el cromosoma 19, codifica una proteína implicada en el metabolismo lipídico, determinando fundamentalmente las concentraciones plasmáticas de LDLc. Las variaciones en la secuencia de este gen originan los alelos E2, E3 y E4, los que dan lugar a cambios de aminoácidos en la proteína en las posiciones 112 y 158. El alelo E2 determina la presencia del aminoácido cisteína en las dos posiciones, el E3 origina una proteína con cisteína en la posición 112 y arginina en la 158, y el alelo E4 posee arginina en ambas posiciones. La frecuencia de estos alelos varía según el origen geográfico de la población: en la población caucásica se ha estimado para E2, 7 %; E3, 78 %, y E4, 15 %. Los portadores del alelo E2 presentan menores concentraciones plasmáticas de LDLc que los homocigotos E3/E3; mientras que los portadores del alelo E4, tienen concentraciones más elevadas de LDLc que los anteriores. Por otro lado, el alelo E4 se ha asociado a un mayor riesgo de enfermedad cardiovascular y de enfermedad de Alzheimer.19 Ante una misma dieta, los individuos con genotipo E2 son hiporrespondedores; los de genotipo E3, son normorrespondedores y los que tienen genotipo E4 son hiperrespondedores.20
Otra proteína con un papel importante en el metabolismo lipídico y el desarrollo de la enfermedad coronaria es la apolipoproteína A1 (APO A1). La sustitución de una guanina por una adenina (A-G) en el promotor del gen APOA1, se asocia con un incremento de colesterol-HDL (alelo B); mientras que el alelo A se relaciona con menores niveles de colesterol-HDL.14,21 Se ha observado que las mujeres que ingieren de forma preferente ácidos grasos poliinsaturados sobre ácidos grasos saturados y monoinsaturados, tienen mayores niveles de HDL. El efecto del tipo de grasa es muy significativo en hombres, sobre todo cuando se considera también el consumo de alcohol y el tabaquismo. Los individuos con partículas de LDL densas y pequeñas (fenotipo B) tienen un mayor riesgo de padecer enfermedades coronarias y son menos respondedores a la dieta baja en grasa que los individuos que muestran partículas de LDL mayores y menos densas (fenotipo A), quienes tienen menor riesgo de enfermedades coronarias. Finalmente existe un fenotipo intermedio, mediado por la dieta, que se comporta como el fenotipo A cuando los individuos siguen una dieta de contenido medio en grasa (32 %), pero como el fenotipo B cuando ingieren menor cantidad de grasa (10 %); este comportamiento puede ser explicado por las interacciones entre el genotipo y la dieta.14,22
Para explicar la etiología de la diabetes mellitus tipo 2 se han expuesto varias teorías: la del genotipo ahorrador, del genotipo no tan ahorrativo, del fenotipo ahorrador y la de comidas genéticamente desconocidas:
1. Teoría del genotipo ahorrador. Trata de dar explicación a la epidemia de diabetes mellitus ocurrida en las comunidades indígenas poscoloniales; plantea que los genes responsables de la resistencia a la insulina protegen a los individuos durante períodos prolongados de ayuno, almacenando la energía en forma de grasa en lugar de glucógeno en el músculo. Este metabolismo ahorrativo permitió la supervivencia de los pobladores migrantes de la última glaciación para utilizar al máximo el tipo de alimentación disponible. Sus descendientes, en el escenario de factores sociales y culturales de la sociedad occidental actual con abundancia de comidas con alto contenido energético y un estilo de vida sedentario, tienen estos genes de "protección" convertidos en "dañinos", sugiriendo que los individuos no están equipados con la maquinaria metabólica para la ingesta abundante. En este genotipo se involucran muchos genes, como los de la adiposidad, que participan en el metabolismo energético y en el riesgo para enfermedades asociadas, entre otros. Se supone que en Europa en el pasado, con un estilo de vida de festejos continuos, disminuyó el genotipo por selección natural de individuos diabéticos, los cuales murieron jóvenes, probablemente por complicaciones de la enfermedad.23
2. Teoría de la resistencia a la insulina como genotipo no tan ahorrativo. Plantea que el elemento más importante que permitió la sobrevida de los pobladores migrantes de la última glaciación durante periodos de hambruna, fue el conservar la masa muscular para poder cazar con éxito, o poder escapar cuando se convertían en presa. Conservar la masa muscular en períodos de hambruna implica reducir la proteólisis en estas células, mecanismo inducido por la resistencia a la insulina. Cuando estos genes son expuestos a la dieta moderna, se desarrolla resistencia a la insulina y posteriormente diabetes mellitus tipo 2. Esta hipótesis de la resistencia a la insulina se apoya en evidencias investigativas en las que se demuestra que el cerebro no necesita aminoácidos como fuente de energía porque es capaz de utilizar beta-hidroxibutirato y acetoacetato en períodos prolongados de ayuno,24 de estos hallazgos se deduce la importancia de los adipocitos involucrados en la obesidad que aparece en los diabéticos. En pacientes con diabetes mellitus tipo II la proteólisis está reducida y no se modifica por administración de insulina. Esta teoría se apoya con el hecho de que el gen CPN10 que codifica la proteína calpaína 10, una proteasa de cisteína que es citosólica, cuando se encuentra en estado homocigoto para el alelo G (GG), produce menor cantidad del ARNm en el músculo estriado de los indios Pima, lo cual provoca una disminución en la proteólisis. Las dos teorías anteriormente mencionadas proponen que las personas con susceptibilidad genética que no desarrollan diabetes mellitus tipo 2, resultan obesas.23
3. Teoría del fenotipo ahorrador. Plantea que una alimentación deficiente en la vida fetal y en la etapa posnatal temprana, activa mecanismos de nutrición ahorrativa; es decir, por acción hormonal se puede modificar la expresión de diferentes genes y la nueva "reprogramación" estará en relación directa con la duración, la etapa del desarrollo y el estado de desnutrición. Un metabolismo ahorrativo puede conducir a la alteración de la estructura y función de las células beta y los islotes de Langerhans, que en algunos casos inducen modificaciones más complejas como reducción en la vasculatura e inervación, que complican aún más la alimentación durante la vida fetal. La producción de insulina se verá afectada según la extensión y el periodo del estado ahorrador. Las personas que tuvieron desnutrición en la etapa fetal quedaron programadas para producir menor cantidad de insulina durante su vida, un cambio brusco a una sobrenutrición en el primer año de vida conduce a diabetes en el futuro por la baja producción de insulina en las células beta y por el metabolismo ahorrativo que posee.23 Esta teoría trata de explicar la diabetes de la población caucásica, en donde se tiene un modo de herencia materna que no se explica por mutaciones en el ADN mitocondrial, esas mujeres diabéticas tienen hijos con bajo peso al nacimiento que en la vida adulta desarrollan enfermedad. En la actualidad se ha dado explicación a tal fenómeno a través del mecanismo epigenético.
4. Teoría de las comidas genéticamente desconocidas. Se basa en que los humanos, antes de descubrir la agricultura como fuente de obtención de alimentos, se alimentaban de frutas y una dieta baja en grasa natural. A medida que fueron modificando esta alimentación se produjo una selección natural. La diabetes mellitus tipo 2 se desarrolla principalmente en poblaciones que no habían modificado de manera importante su alimentación; sin embargo, consumen bebidas sin potasio y con mayor concentración de azúcar refinada (que es genéticamente desconocida o, lo que es lo mismo, no disponen de genes para el metabolismo de este nuevo producto que se les presenta), así como grasa en cantidades superiores a la capacidad del metabolismo (cantidades genéticamente desconocidas). Cuando se analiza la prevalencia de diabetes en europeos, se estima que pudieron ser eliminados gradualmente los diabéticos, de ahí la baja prevalencia en esos países.23,25
Se han obtenido otras evidencias al respecto:25
· Al analizar la importancia de la combinación de la sacarosa con el potasio en cortadores de caña; estos hombres consumían gran cantidad de azúcar al masticar caña y sin embargo no presentaban diabetes, en cambio sus patrones, que consumían azúcar refinada, sí presentaban la enfermedad.
· Estudios realizados en grupos étnicos contemporáneos con elevada prevalencia de diabetes mellitus tipo 2, en los que el consumo de azúcar refinado se ha incrementado al incluirlo como edulcorante en Coca Cola, hot-cakes, gelatinas artificiales, pan dulce industrial, helados de crema, leche condensada y otras confituras, y que ahora forman parte de su dieta.
· En los indios Pima, el consumo de grasa antes de la epidemia de diabetes era del 8-12 %, ahora representa el 50 % de su dieta.
· Un grupo de aborígenes australianos mejoraron la obesidad y la diabetes tipo 2 en solo 5 semanas al regresar a su dieta tradicional baja en grasa.
Utilizando marcadores genéticos polimórficos se ha demostrado que los genes asociados a diabetes mellitus tipo 2 no están combinados de la misma forma en las poblaciones; por ejemplo: la asociación del locus D2S125 en la población méxico-americana y no en la población finlandesa, en la que es más frecuente el locus D22S426.23
Con relación al cáncer, la metilenotetrahidrofolato reductasa (MTHFR) es una proteína importante en las reacciones de metilación. El polimorfismo C667T (alanina a valina) en el gen MTHFR, produce una reducción de la actividad enzimática y está asociado inversamente con la presencia de cáncer colorrectal y leucemia linfocitaria aguda. Una ingesta baja de folato, vitamina B 12, vitamina B6 o metionina se asocia también con un mayor riesgo de cáncer entre los que poseen el genotipo MTHFR TT.14
NUTRIGENÓMICA
Esta rama de la Genómica nutricional se dedica a estudiar cómo lo que comemos puede modificar la expresión de los genes; es decir, cómo los componentes de la dieta pueden actuar sobre el genoma humano, directa o indirectamente, y pueden alterar la expresión o estructura de los genes. De este modo puede decirse que, bajo ciertas circunstancias, la dieta puede ser un factor de riesgo de una enfermedad.26,27
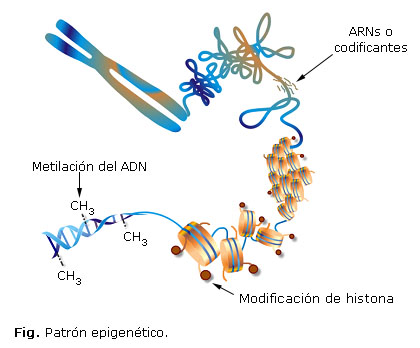
El mecanismo epigenético explica de qué manera los alimentos pueden alterar la expresión de los genes, modificando el patrón epigenético. El epigenoma está conformado por todas aquellas moléculas que se asocian al ADN en su enrollamiento, las cuales modifican su expresión sin modificar la secuencia de nucleótidos (Fig.). La estructura de la cromatina es un mecanismo de control de la transcripción, debido al elevado nivel de compactación del genoma; por lo que se necesita un cambio conformacional en dicha estructura para permitir la expresión genética. Por lo que podemos decir, que esta última depende en gran medida de la capacidad de remodelamiento de la cromatina, tanto a nivel de las regiones reguladoras como de sus secuencias codificantes y no codificantes.26
Los principales mecanismos que participan en la regulación epigenética a nivel de la cromatina son básicamente tres: metilación del ADN, modificaciones postraduccionales de las histonas (acetilación y desacetilación), y los microARN.28,29 Existe un cuarto mecanismo al que se hace referencia que es la impronta genómica, la cual está estrechamente ligada a la metilación del ADN. En esta revisión abordaremos los dos primeros mecanismos pues, al parecer, son los más relacionados con la dieta.
La metilación del ADN consiste en la inserción de un grupo metilo (CH3) en la posición 5 de la base nitrogenada dioxicitosina (dC) para formar 5-metilcitosina. La reacción es catalizada por la enzima DNA metiltransferasa (DNMTs), en presencia de un sustrato donador de grupos metilo (S-adenosilmetionina). La 5-metilcitosina se encuentra en los dinucleótidos citosina-guanina (CpG), los que no presentan una distribución uniforme en el genoma humano. En el 98 % del genoma, los CpG están presentes, en promedio, una vez por cada 80 dinucleótidos; sin embargo, existen regiones de 200 pares de bases en las que la frecuencia de dinucleótidos CpG es 5 veces mayor y se les denomina islotes CpG. Estos se localizan entre la región central del promotor y el sitio de inicio de la transcripción, observándose represión en la expresión del gen cuando se encuentran metiladas.26,28,29
Las histonas son proteínas básicas presentes en el núcleo celular que se encargan de empaquetar el ADN formando el nucleosoma (octámero de histonas, sobre el que la molécula de ADN da dos vueltas). Las histonas que conforman esta estructura son: H2A, H2B, H3 y H4 (dos de cada una de ellas). El conjunto de nucleosomas continúa enrollándose para formar la cromatina, cuyo grado máximo de empaquetamiento está representado por la estructura de cromosoma. La carga básica (positiva) de las histonas, les facilita unirse al ADN. Estas proteínas sufren modificaciones postraduccionales de acetilación y desacetilación, lo cual puede alterar sus propiedades de unión al ADN y produce modificación de la estructura de la cromatina. Asimismo, existe una correlación entre la acetilación de histonas (especialmente de las histonas H3 y H4) y el incremento de la transcripción génica: al acetilarse, la histona posee menor afinidad hacia la molécula de ADN, por cambio en la carga electrostática de la proteína, lo que produce una menor compactación de la cromatina, dando por resultado que los factores de transcripción reconozcan más fácilmente al promotor de los genes y se produzca la transcripción. La desacetilación de las histonas, por el contrario, origina una configuración más compacta de la cromatina, que se traduce en una inhibición de la transcripción.
Las reacciones químicas descritas anteriormente consisten, en esencia, en el cambio de un grupo amino terminal (NH3) de los residuos de lisina en las regiones amino terminales de las histonas H3 y H 4 por un grupo acetilo (COCH3), lo que neutraliza la carga positiva de las histonas y permite que el ADN se exponga a los factores de transcripción. Estas reacciones son catalizadas por las acetiltransferasas. Al concluir la transcripción es necesario que el ADN y las histonas vuelvan a empaquetarse, proceso que se realiza por la acción de las enzimas desacetilasas de histonas.
En la actualidad existe gran interés en conocer y caracterizar qué alimentos tienen efectos en la regulación epigenética. Recientemente se han reportado algunos alimentos que contienen sustancias que actúan como inhibidores de las desacetilasas de histonas, lo que ha adquirido gran importancia en el campo de la oncología, como alternativa preventiva y terapéutica. Por lo tanto, aquellos alimentos que tienen sustancias con esta función se están investigando, especialmente para prevenir y tratar cáncer de mama, colon, estómago y próstata, debido a sus propiedades para permitir la expresión de genes silenciados epigenéticamente que están involucrados en la regulación del ciclo celular, apoptosis y/o mecanismos de destoxificación.12,26
Los polifenoles son sustancias presentes en frutas y vegetales y constituyen una parte importante de la dieta. Pueden dividirse, al menos, en 7 clases diferentes según su estructura química: flavonoides, estilbenos, ácidos fenólicos, benzoquinonas, acetofenonas, ligninas y xantonas.
Algunos estudios estiman que existen hasta 8 000 polifenoles diferentes en la dieta, entre ellos la epigalotocatecina-3-galato (EGCG) que se encuentra con abundancia en el té verde; la curcumina en el curry; el resveratrol presente en las uvas y el vino tinto; la genisteína en la soya, y el selenio en la cebolla y las nueces brasileñas. Estos compuestos, debido a su capacidad de alterar mecanismos epigenéticos mediante la remodelación de la cromatina y activación de genes silenciados, por su capacidad de modificar la acetilación y desacetilación de histonas e inhibir DNMTs, tienen una participación e impacto significativos en la prevención del cáncer.26,30,31
Diversos estudios han demostrado que otras sustancias como el sulforafano, alil-mercaptano, dialil-disulfuro y butirato contenidas en distintos alimentos, a través de mecanismos aún desconocidos, pueden regular la metilación del ADN y la acetilación y desacetilación de las histonas.32 El sulforafano (1-isotiocianato-4-[metilsulfinil]-butano; CH3-SO-(CH2)4-N = C = S) es un miembro de la familia de isotiocianatos y se encuentra presente en vegetales como el brócoli y la col. Ha despertado gran atención por sus propiedades anticancerígenas, además de sus efectos antimicrobianos; este ha mostrado una protección significativa contra el cáncer de mama inducido químicamente con el agente 9,10 dimetil-1,2 benzantraceno en ratas y contra el cáncer de estómago inducido con benzo[a]pireno en ratones. El mecanismo sugerido por el cual el sulforafano protege contra el cáncer es a través de la inducción de la expresión de enzimas destoxificantes como la glutatión transferasa.26,33 Otros investigadores demostraron que el tratamiento de cáncer de colon humano de células HT29, con sulforafano, genera la detención del ciclo celular en la fase G2-M e induce apoptosis. El mecanismo a través del cual el sulforafano logró tales efectos se asocia a su propiedad para actuar como inhibidor de las desacetilasas de histonas y permitir la expresión del gen BAX, el cual codifica una proteína que favorece la apoptosis.34
El dialil-disulfuro y el alil-mercaptano (un metabolito de la S-alil-mercaptocisteína) son compuestos organosulfurados que se encuentran en los ajos y las cebollas. Algunos estudios han mostrado que esos compuestos tienen efectos antiproliferativos e inducen apoptosis por el mecanismo de la hiperacetilación de las histonas, sugiriendo que pueden actuar como inhibidores de las desacetilasas de histonas. Diversos estudios han analizado estos compuestos in vitro e in vivo e identificaron que el alil-mercaptano es el inhibidor competitivo más potente de la actividad de las desacetilasas de histonas entre los compuestos órganosulfurados estudiados.35,36 De tal forma, en el cáncer de colon, el alil-mercaptano induce la acumulación de histonas acetiladas y promueve la unión del factor transcripcional Sp3 a la región promotora del gen P21WAF1, incrementando su expresión a nivel de ARNm y de la proteína resultante de dicho gen; el efecto es el bloqueo del ciclo celular y la inhibición de la proliferación de células tumorales.35
Actualmente la Nutrigenómica intenta dilucidar en qué concentraciones todos estos componentes de los alimentos logran los efectos referidos en las células tumorales, así como el tiempo debe durar la administración de estos.
CONCLUSIONES
La apertura de esta nueva rama de la investigación médica y los conocimientos que ha aportado crean nuevas expectativas sobre la prevención y el tratamiento de enfermedades complejas; y plantean que los estilos de vida saludables en estrecho equilibrio con el ambiente son los compatibles con un estado de salud plena. Estos pronunciamientos sirven de base para recomendar una dieta equilibrada, una actividad física regular, el mantenimiento de estados emocionales positivos y evitar la exposición a sustancias químicas que pueden alterar la expresión de nuestro genoma y, por tanto, nuestro estado de salud.
REFERENCIAS BIBLIOGRÁFICAS
1. Camp KM, Trujillo E. Position of the Academy of Nutrition and Dietetics: nutritional genomics. J Acad Nutr Diet. 2014;114:299-312.
2. Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256-60.
3. Ordovás JM, Corella D. La revolución del genoma humano. ¿Qué significa genómica, epigenética, nutrigenómica, metabolómica? En: Arola L, Arroyo E, Baiges I, Bermejo M, Boada J, Belmonte S, et al. Genética, nutrición y enfermedad. [Internet]. Madrid: EDIMSA; 2008 [citado 20 Oct 2016]. 17 p. Disponible en: http://www.genutren.es/actividades/LibroGenutren.pdf
4. Xacur GF, Castillo QJI, Hernández EVM, Laviada MH. Genómica nutricional: una aproximación de la interacción genoma-ambiente. Rev Méd Chile. 2008;136:1460-7.
5. Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848-53.
6. Hernández FR. Enfermedades causadas por mutaciones monogénicas. En: Lantigua CA. Introducción a la Genética Médica. 2a ed. La Habana: Ecimed; 2011. p. 179-208.
7. Lantigua CA. Introducción a la Genética Médica. 2da. ed. La Habana: Ecimed; 2011. p. 287-305.
8. Hernández CY, Lardoeyt FR, Rosado RAI, Martínez SA. Interacción del genoma y el ambiente en la aparición de la diabetes mellitus tipo 2 en una población del municipio Jaruco, 2008-2009. Rev Cubana Genet Comunit. 2012;6(1):31-9.
9. Torres SY, Lardoeyt FR, Peláez LC, Marcheco TB. Contribución de los factores genéticos y ambientales en la aparición de la hipertensión arterial a través de un estudio de gemelos. Rev Cubana Genet Comunit. 2008;2(2):52-8.
10. Hernández FRA. Genoma y ambiente en la génesis de la obesidad. Rev Cubana Genet Comunit. 2013;7(1). Disponible en: http://www.sld.cu/revistas/rcgc/v7n1/rcgc010113.html [Citado junio 2015].
11. Sales P, Pelegrini B, Goerch MC. Nutrigenomics: Definitions and Advances of this New Science. J Nutr Metab. 2014;20:27-59.
12. De Lorenzo D. Perspectivas presentes y futuras de la Nutrigenómica y la Nutrigenética en la medicina preventiva. Nutr Clin Diet Hosp. 2012;32(2):92-105.
13. Sanhueza CJ, Valenzuela BA. Nutrigenómica: revelando los aspectos moleculares de una nutrición personalizada. Rev Chil Nutr. 2012;39(1):71-85.
14. Marti A, Moreno AMJ, Zulet A, Martínez JA. Avances en nutrición molecular: nutrigenómica y/o nutrigenética. Nutr Hosp. 2005;20(3):157-64.
15. Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Natura. 1998;392:398-401.
16. Corella D, Portolés O. Avances en el conocimiento de las bases genéticas de la obesidad. En: Arola L, Arroyo E, Baiges I, Bermejo M, Boada J, Belmonte S, et al. Genética, nutrición y enfermedad. [Internet]. Madrid: EDIMSA; 2008 [citado 20 oct 2016]. 51p. Disponible en: http://www.genutren.es/actividades/LibroGenutren.pdf
17. Martí A, Corbalan MC, Forga L, Martínez JA, Hinney A, Hebebrand J. Presence of new mutation in the melanocortin-4 receptor in a Spanish population. Int J Obes Relat Metab Disor. 2003;27:385-8.
18. Quiroga MI. Hipertensión arterial. Aspectos genéticos. An Fac Med. 2010;71(4):231-5.
19. Corella D, Tucker K, Lahoz C. Alcohol drinking determines the effect of the APOE locus on LDL-cholesterol concentrations in men: the Framingham Offspring Study. Am J Clin Nutr. 2001;73:736-45.
20. Universidad de Barcelona. Facultad de Farmacia. Nuevos avances en nutrición: Nutrigenética y Nutrigenómica. 6a Reunión de la Sociedad Española de Seguridad Alimentaria. 2009. Disponible en: http://www.sesal.org [Revisado junio 2015]
21. Ordovás JM. HDL genetics: candidate genes, genome wide scans and gene-environment interactions. Cardiovasc Drugs Ther. 2002;16:273-81.
22. Krauss RM. Dietary and genetic effects of LDL heterogeneity. World Rev Nutr Diet. 2001;89:12-22.
23. Carrillo C, Panduro CA. Genética de la diabetes mellitus tipo 2. Investigación en salud. 2001;3(99):27-34. Disponible en: http://www.redalyc.org/articulo.oa?id=14239905 [Revisado octubre 2014]
24. Owen OE, Morgan AP, Kemp HG, Sullivann JM, Herrera MG, Cahill GF Jr. Brain metabolism during fasting. J Clin Invest. 1967;46:1589-97.
25. Baschetti R. Diabetes epidemic in newly westernized populations: is it due to thrifty genes or to genetically unknown foods? J R Soc Med. 1998;91:622-5.
26. Martínez LE, García GMR, Campos PWY, González BK. Genómica nutricional: conceptos y perspectivas. Rev Endocrinol Nutr. 2013;21(1):22-34. Disponible en: http://www.medigraphic.com/endocrinologia [Revisado julio 2015]
27. McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health. Acta Physiol (Oxf). 2011;202(2):103-18.
28. Bedregal P, Shand B, Santos MJ, Ventura-Juncá P. Aportes de la epigenética en la comprensión del desarrollo del ser humano. Rev Med Chile. 2010;138:366-72.
29. García-Giménez JL. Epigenética. La gramática del código genético. Journal of Feelsynapsis. 2012;4:34-8.
30. Vargas HJE, Camacho GMP, Ramírez PD. Efectos de los nutrientes y compuestos bioactivos de los alimentos en tejidos y células de cáncer humano: aproximación nutrigenómica. Rev Fac Med. 2013;61(3):293-300.
31. Villegas VCA, Faxas GME. La nutrición en la inmunidad y el cáncer. Rev Argent Endocrinol Metab. 2014;5(1):30-6.
32. Hardy TM, Tollefsbol TO. Epigenetic diet: impact on the epigenome. Epigenetics. 2011;503-18.
33. Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem Pharmacol. 2013;85:1398-404.
34. Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Research. 2000;60:1426-33.
35. Nian H, Delage B, Pinto JT, Daswood RH. Alyll mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the p21WAF1 promoter. Carcinogenesis. 2008;29:1816-24.
36. Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drugs Metabolism. 2000;32:395-411.
Recibido: 4 de
marzo de 2016.
Aprobado:
20 de junio de 2016.
Tamara Rubio González. Universidad de Ciencias Médicas de Santiago de Cuba. Correo electrónico: trubio@medired.scu.sld.cu