PRESENTACIÓN
DE CASO
Múltiples
afecciones en mujer de 75 años
Multiple conditions
in a 75 year-old woman
Dr. Cosme M.
Cand Huerta, Dr. Carlos A. Domínguez Álvarez.
Hospital Clínico
Quirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
Se presenta una
mujer de 75 años que ingresó por fiebre, visceromegalia, deficiencia
inmunológica con pancitopenia e hipogammaglobulinemia asociada a cifras
milenarias de deshidrogenasa láctica y ferritina. Se aplicaron los criterios
diagnósticos del síndrome hemofagocítico. Se realiza revisión
de la literatura.
Palabras clave:
síndrome hemofagocítico, deficiencia inmunológica.
ABSTRACT
A case of a 75
year-old woman is presented because she was admitted due to fever, visceromegaly,
immune deficiency with pancytopenia and hypogammaglobulinemia associated with
ancient figures of ferritin and lactic dehydrogenase. Hemophagocytic syndrome
diagnostic criteria were applied. Literature review is performed.
Keywords:
hemophagocytic syndrome, immune deficiency.
INTRODUCCIÓN
La linfohistiocitosis
hemofagocítica es un síndrome hiperinflamatorio caracterizado por
una excesiva función de los macrófagos. Este trastorno, aunque ligado
a específicas anomalías de orden genético en niños, por
notables mutaciones del gen perforina, ocurre en adultos de manera esporádica.
Aquí se presenta, en forma de discusión diagnóstica, una pacinete
con una constelación clínica y analítica que cumple los criterios
de la entidad y cuyo diagnóstico etiológico se confirmó por el
estudio de médula ósea.
PRESENTACIÓN
DEL CASO
Mujer de 75 años
de edad que ingresó por referir dolores óseos generalizados, a predominio
de la región pélvica y muslos, que aumentaban con los esfuerzos y
aliviaban con el reposo y con dosis bajas de antiinflamatorios no esteroideos,
de dos meses de evolución además de presentar una fiebre diaria de
38 grados, generalmente vespertina precedida de escalofríos, durante la
última semana.Trajo de consulta externa cifras bajas de hemoglobina y eritrosedimentación
centenaria.
Antecedentes
patológicos personales : alergia al yodo. No transfusiones, operaciones
ni hábitos tóxicos.
Examen físico
: mucosas hipocoloreadas. Índice de masa corporal en 23,6 kg/m2. TA 130
con 80 mm Hg, FC 92 por minuto, FR 14 por minuto, temperatura 38,3 grados Celsius.
A nivel de las articulaciones interfalángicas distales de ambas manos había
nódulos no dolorosos tipo Heberden y la movilización de pelvis y miembros
inferiores era dolorosa aunque la maniobra de Lasegue fue negativa. El abdomen
era depresible e indoloro en donde se palpaba hepatomegalia lisa de borde fino
y no dolorosa rebasando 2 cm. El reborde costal con borde superior en segundo
espacio intercostal y altura hepática de 14 cm a nivel de línea medio
clavicular. Existía matidez en el 9no espacio intercostal izquierdo a nivel
de la línea axilar anterior pero no se palpaba el bazo. El resto del examen
fue negativo.
Evolución
médica y complementaria
Los estudios analíticos
se muestran en la tabla 1. La enferma comenzó a presentar
melenas y se realizó endoscopia superior que reveló múltiples
úlceras gástricas con signo de sangrado reciente pero no activo y
esofagitis micótica grado II, por lo cual se inició tratamiento antiulceroso,
nistatina oral y transfusión de dos unidades de glóbulos rojos. Evolutivamente,
la enferma continuó con picos febriles vespertinos diarios y se realizó
una serie de estudios imaginológicos cuyos resultados aparecen en la tabla
2.
|
Tabla 1. Resultados de Laboratorio
|
|
Exámenes
|
Valor Normal
|
Resultado
|
|
Hemoglobina
|
120-160 g/l
|
70
|
|
Vol.corpusc.
medio (VCM)
|
80-100 fl
|
83
|
|
Eritrosedimentación
|
0-20mm/h
|
110
|
|
Leucocitos
|
4-11x109/l
|
2,9
|
|
Neutrófilos
|
50-70 %
|
49,5
|
|
Linfocitos
|
20-40 %
|
36,8
|
|
Monocitos
|
2-10 %
|
13,7
|
|
Glucemia
|
4,2-6,1 mmol/l
|
4,7
|
|
Creatinina
|
47-113,3 mcmol/l
|
80
|
|
Urea
|
1,7-8,3 mmol/l
|
7,54
|
|
Proteínas Totales
|
60-80 g/l
|
57,4
|
|
Albúmina
|
34-50 g/l
|
23,2
|
|
IgG
|
8-17 g/l
|
5,60
|
|
IgA
|
0,65-4,9 g/l
|
0,636
|
|
IgM
|
0,5-2,7 g/l
|
0,203
|
|
ASAT
|
0-40 U/l
|
42,3
|
|
ALAT
|
0-40 U/l
|
48,5
|
|
Bilirrubina
Total
|
0-17 mcmol/l
|
6,9
|
|
Fosfatasa alcalina
|
35-130 U/l
|
182
|
|
Deshidrog.
Láctica(LDH)
|
135-250 U/l
|
7081
|
|
Ferritina
|
12-400 ng/ml
|
15440
|
|
Haptoglobina
|
0,2-2 g/l
|
2,97
|
|
Proteína C
reactiva
|
0-5 mg/l
|
175,10
|
|
Ácido Úrico
|
142-416,4 mcmol/l
|
259
|
|
Calcio Sérico
|
2,02-2,6 mmol/l
|
2,18
|
|
Gammaglutamil
transpeptidasa
|
7-50 U/l
|
45
|
|
Colesteroles
|
3,6 -5,2 mmol/l
|
4,44
|
|
Triglicéridos
|
0,5- 1,85 mmol/l
|
8,08
|
|
Creatinquinasa
|
0-170 U/l
|
169
|
|
Hemocultivos
|
negativos
|
tres negativos
|
|
Amilasa sérica
|
0-100 UI
|
54,8
|
|
Test de Coombs
|
directo e indirecto
negativos
|
negativos
|
|
Hierro Sérico
|
6,6-26 mmol/l
|
4,7
|
|
Dímero D
|
Negativo
|
positivo >0,5mcg/ml
|
|
Anticuerpo
para HIV
|
Negativo
|
negativo
|
|
Serología CMV
IgM
|
Negativo
|
negativo
|
|
Serología EBV
IgM
|
Negativo
|
negativo
|
|
Plaquetas
|
150-400 x 10
9/l
|
50
|
|
Sodio
|
135-145 mEq/l
|
140,8
|
|
Potasio
|
3,5-5 mEq/l
|
4,37
|
|
Cloro
|
99-110 mEq/l
|
105,2
|
DISCUSIÓN
CLÍNICA
Llamó la
atención la importante afectación inmunológica (que explicaba
la fiebre de 2 meses de evolución) y la pancitopenia periférica. Las
inmunodeficiencias primarias que más afectan a los adultos no podían
ser planteadas como son la deficiencia selectiva de IgA, la relacionada con
globulinas séricas normales, la ligada al cromosoma X y la asociada a un
timoma o síndrome de Good porque existía depresión severa de
todos los tipos de inmunoglobulinas, por la edad avanzada de la enferma y porque
no había evidencia alguna de enfermedad tímica. Otra entidad que se
tuvo en cuenta fue la inmunodeficiencia variable común, de la cual se tiene
experiencia en nuestro servicio, pero la ausencia del típico aumento de
la susceptibilidad para infecciones piógenas dada por las frecuentes sepsis
respiratorias y las diarreas malabsortivas nos hizo descartar esta frecuente
deficiencia de la inmunidad humoral. Resta entonces discutir los trastornos
inmunológicos secundarios donde no hay cabida para el síndrome de
inmunodeficiencia adquirida o SIDA, esplenectomía, fármacos inmunosupresores,
desnutrición ni hepatopatías crónicas dado los estudios complementarios
realizados y la ausencia de antecedentes. Una causa frecuente en la edad de
la enferma era el cáncer de estómago y las neoplasias hematológicas
malignas, éstas últimas, mucho más probables que el primero por
la ausencia de síntomas digestivos y por la mayor relación con la
pancitopenia periférica.
El trastorno sanguíneo
antes mencionado, la normalidad del VCM y la haptoglobina y las indiscutibles
evidencias clínicas hicieron pensar básicamente en un síndrome
mielodisplástico, un linfoma maligno o una rara leucosis aleucémica.
Faltaban en esta paciente las típicas linfadenopatías gomosas y aunque
presentaba adenopatías peripancreáticas, sus características
imaginológicas eran inflamatorias y no linfomatosas, no obstante, la LDH
favorecía a un proceso linfoproliferativo aunque era muy llamativa su cifra
milenaria. La mielodisplasia era probable, aunque se negaba estudios hematológicos
previos y recientes anormales, ni tenía un VCM elevado. Sin embargo, sabíamos
que 30 % de los síndromes mielodisplásicos evolucionan a una leucosis
aguda que en este caso también era probable. Lógicamente se solicitó
lámina periférica con los siguientes resultados: blastos 7 %, mielocitos
5 %, linfocitos 51 % y neutrófilos 30 % y se le practicó una biopsia
de médula ósea ante la sospecha de una leucemia linfoide aguda. Se
decidió de común acuerdo con el servicio de hematología asociar
al tratamiento IV con cefepime, sulfaprim y metronidazol, prednisolona endovenosa
en altas dosis además de albúmina, transfusiones de glóbulos
rojos y concentrado de plaquetas.
Mientras tanto
quedaban otros parámetros analíticos que eran dignos de meditación
como la LDH en más de 7000 U/l, los triglicéridos en 8,08 mmol/l y
muy particularmente, la cifra de ferritina en más de 15000 ng/ml lo que
nos llevó a sospechar el diagnóstico de una linfohistiocitosis hemofagocítica
o síndrome hemofagocítico (LHH). Las otras causas de hiperferritinemia
de tal magnitud, como la histoplasmosis generalizada y la enfermedad de Still
del adulto,1 no eran planteables.
Al aplicar los
criterios diagnósticos originales de Henter (tabla
3):
§ Se necesitan
5 de los 8 criterios para establecer el diagnóstico.
Se comprobó
que el cuadro clínico de la paciente satisfacía los criterios mínimos
para el diagnóstico sospechado al quedar solamente la demostración
convincente de la presencia de la hemofagocitosis en la biopsia de médula
ósea pero la enferma falleció antes de la confirmación histológica.
Igualmente, se aplicaron otros criterios como el de Gritta Janka,3
semejante al arriba citado y también al de Alexandra H. Filipovich4
como sigue en la tabla 4:

Al plantearnos
una LHH en aquel momento, se recalcaba una complicación hiperinflamatoria
altamente letal con hasta 95 % de mortalidad según la causa subyacente
y aunque nuestra paciente no mostró síntomas del sistema nervioso
central como parálisis de nervios craneales y convulsiones, algunos autores
reportan que son comunes. En lo que todos coinciden es que se trata de una abrumadora
inflamación debida a la proliferación e hiperactivación descontroladamente
agresiva de macrófagos bien diferenciados que secretan grandes cantidades
de citoquinas inflamatorias5 y que son capaces de fagocitar otras
células sanguíneas aunque no hace mucho se postuló que la citopenia
es más por mielosupresión que por la gula linfohistiocítica.6
En la práctica
clínica, el mayor peso específico corresponde al principio de que
no se trata de una enfermedad única, sino que responde a una gama de situaciones,
tanto genéticas; como por ejemplo la variante familiar de Farquhar y las
asociadas a inmunodeficiencias (Chédiak-Higashi y el síndrome linfoproliferativo
ligado al cromosoma X); como adquiridas donde se incluyen múltiples enfermedades
infecciosas bacterianas, virales, micóticas, parasitarias y por rickettsias7
así como neoplasias, enfermedades autoinmunes como el lupus8
y exposición a fármacos inmunomoduladores. En nuestra enferma no pudimos
comprobar virosis ni sepsis bacterianas pero los procesos malignos, en particular,
la leucemia linfoide aguda, los linfomas malignos y la mielodisplasia fueron
diariamente analizados en sala (ver discusión arriba). La severidad del
síndrome hemofagocítico hizo perentorio el control inmediato de los
siguientes perfiles (a) etiológico sobre todo infeccioso, (b) fisiopatológico
al controlar la excesiva función de los macrófagos y la tormenta de
citoquinas 9,10 y (c) medidas de sostén. Por lo tanto, se impuso
la supresión inmunológica con el inicio inmediato de glucocorticoides
por su rápida acción y eficacia en tanto la globulina inmune fue solicitada
como buen agente modulador en casos de sepsis más cobertura antibiótica11.
Aunque los agentes infecciosos más frecuente son los virus de Ebstein-Barr
y de la inmunodeficiencia humana, en el caso que aquí se discutió,
no se comprobó positividad microbiológica ni serológica.
DIAGNÓSTICO
CLÍNICO
Inmunodeficiencia
secundaria y linfohistiocitosis hemofagocítica secundaria a enfermedad
hematológica maligna. Sospecha de leucemia linfoblástica aguda.
DISCUSIÓN
PATOLÓGICA
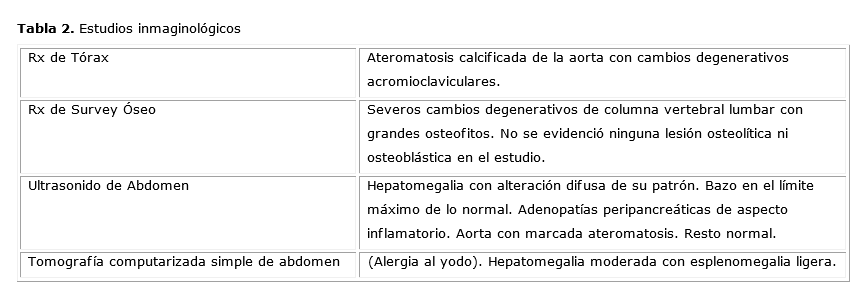
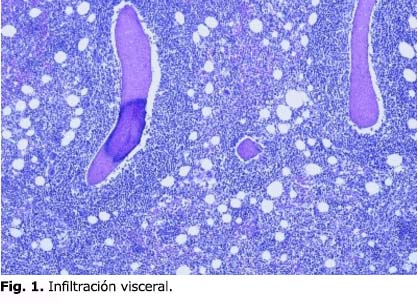
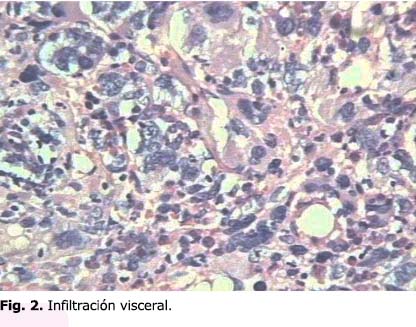
En la autopsia,
los hallazgos más sobresalientes no estuvieron relacionados con las complicaciones
de la inmunosupresión ni la sepsis generalizada, sino con la infiltración
visceral a consecuencia de una leucemia linfoblástica aguda (fig.
1 y 2) en tanto la médula ósea reveló
numerosos histiocitos al contener eritrocitos fagocitados (fig.3).
DIAGNÓSTICO
ANATÓMICO
Leucemia linfocítica
aguda en crisis blástica. Linfohistiocitosis hemofagocítica secundaria.
REFERENCIAS
BIBLIOGRÁFICAS
1. Tierney LM,
Thabet A, Nishino H. Case Records of the Massachusetts General Hospital (Case
10-2011): A woman with fever, confusion, liver failure, anemia and thrombocytopenia.
N Engl J Med. 2011;364:1259-70.
2. Henter JI,
Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis.
Semin Oncol. 1991;18:29-33.
3. Janka G, Zur
Stadt U. Familial and acquired hemophagocytic lympho - histiocytosis. Hematology
Am Soc Hematol Educ Program. 2005:82-8.
4. Filipovich
AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders.
Hematology Am Soc Hematol Educ Program. 2009;17:127-31.
5. Berkun Y, Padeh
H. Macrophage activation syndrome in juvenile idiopathic Arthritis. In: Shoenfeld
Y, Cervera R, Gershwin ME. Diagnostic Criteria in Autoimmune Diseases. USA:
Edit. Humana Press. 2008:21-4.
6. Deane S, Selmi
C, Teuber SS, Gershwin ME. Macrophage activation syndrome in autoimmune disease.
Int Arch Allergy Immunol. 2010;153:109-20.
7. Maakaroun NR,
Moanna A, Jacob JT, Albrecht H. Viral infections associated with hemophagocytic
syndrome. Rev Med Virol. 2010;20:93-105.
8. Uttenthal BJ,
Layton DM, Vyse TJ, Schreiber BE. Clinical problem solving: The wolf at the
door. N Engl J Med. 2012;366:2216-21.
9. Fisman DN.
Hemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6:601-8.
10. Freeman HR,
Ramanan AV. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96:688-93.
11. Shabbir M,
Lucas J, Lazarchick J, Shirai K. Secondary hemophagocytic syndrome
in adults: a case series of 18 patients in a single institution and review
of literature. Hematol Oncol. 2011;29:100-6.
Recibido: 8 de
Julio de 2014.
Aprobado:
30 de Julio de 2014
Haydée
del Pozo
Correo electrónico:hadpojez@infomed.sld.cu
{kind=link}